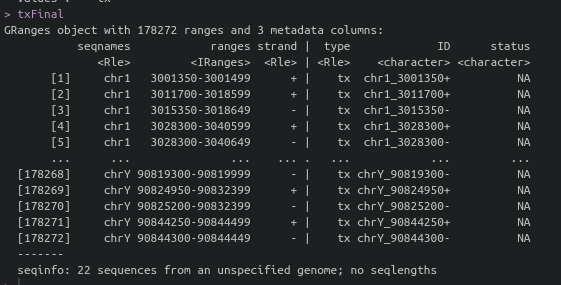
I'm trying to process PRO-seq data using groHMM. I tried to follow tutorial but I think annotation was not performed well. I could not find gene annotation information in txFinal object.
My sample is for mouse so I downloaded mm10 known gene. For processing, there was no error or warning messages.
I realized when I tried to perform DE analysis using edgeR. Before I make count matrix from txFinal object, I realized there is not any annotated gene information. I attached my code below. If I misunderstand or miss something about whole process, let me know.
### Transcript Calling ###
Sall <- sort(c(Control,Case))
Sall
hmmResult <- detectTranscripts(Sall,LtProbB = -200,UTS = 5,
threshold = 1)
txHMM <- hmmResult$transcripts
### Evaluation transcript calling ###
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
kgdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
kgdb
library(GenomicFeatures)
kgAll <- transcripts(kgdb, columns=c("gene_id","tx_id","tx_name"))
seqlevels(kgAll) <- seqlevelsInUse(kgAll)
kgConsensus <- makeConsensusAnnotations(kgAll,keytype="gene_id",mc.cores=getOption("mc.cores"))
library(org.Mm.eg.db)
map <- select(org.Mm.eg.db,keys=unlist(mcols(kgConsensus)$gene_id),columns=c("SYMBOL"), keytype=c("ENTREZID"))
mcols(kgConsensus)$symbol <- map$SYMBOL
mcols(kgConsensus)$type <- "gene"
e <- evaluateHMMInAnnotations(txHMM,kgConsensus)
e$eval
### HMM tuning ###
tune <- data.frame(LtProbB=c(rep(-100,3),rep(-200,3),rep(-300,3)),
UTS=rep(c(5,10,15),3))
Fp <- windowAnalysis(Sall, strand="+", windowSize=50)
Fm <- windowAnalysis(Sall, strand="-", windowSize=50)
evals <- mclapply(seq_len(9), function(x) {
hmm <- detectTranscripts(Fp=Fp, Fm=Fm, LtProbB=tune$LtProbB[x],
UTS=tune$UTS[x])
e <- evaluateHMMInAnnotations(hmm$transcripts, kgConsensus)
e$eval
}, mc.cores=getOption("mc.cores"), mc.silent=TRUE)
tune <- cbind(tune, do.call(rbind, evals)) # evals from the previous run
tune
getExpressedAnnotations <- function(features, reads) {
fLimit <- limitToXkb(features)
count <- countOverlaps(fLimit, reads)
features <- features[count!=0,]
return(features[(quantile(width(features), .05) < width(features))
& (width(features) < quantile(width(features), .95)),])}
conExpressed <- getExpressedAnnotations(features=kgConsensus,reads=Sall)
conExpressed@ranges
td <- getTxDensity(txHMM, conExpressed, mc.cores=getOption("mc.cores"))
u <- par("usr")
lines(c(u[1], 0, 0, 1000, 1000, u[2]), c(0,0,u[4]-.04,u[4]-.04,0,0),col="red")
legend("topright", lty=c(1,1), col=c(2,1), c("ideal", "groHMM"))
text(c(-500,500), c(.05,.5), c("FivePrimeFP", "TP"))
td
bPlus <- breakTranscriptsOnGenes(txHMM, kgConsensus, strand="+")
bMinus <- breakTranscriptsOnGenes(txHMM, kgConsensus, strand="-")
txBroken <- c(bPlus, bMinus)
txFinal <- combineTranscripts(txBroken, kgConsensus)
tdFinal <- getTxDensity(txFinal, conExpressed, mc.cores=getOption("mc.cores"))
Hello :)
I'm trying to process PRO-seq data using groHMM. I tried to follow tutorial but I think annotation was not performed well. I could not find gene annotation information in txFinal object.

My sample is for mouse so I downloaded mm10 known gene. For processing, there was no error or warning messages.
I realized when I tried to perform DE analysis using edgeR. Before I make count matrix from txFinal object, I realized there is not any annotated gene information. I attached my code below. If I misunderstand or miss something about whole process, let me know.
Thanks!!