From 457a0d13e1946210f32975527c98f489fa73ff4a Mon Sep 17 00:00:00 2001
From: YB_HU <138468445+RussellHu41@users.noreply.github.com>
Date: Wed, 17 Jun 2026 11:53:03 +0800
Subject: [PATCH] Update DeePMD_2026_05_19.md
---
source/_posts/DeePMD_2026_05_19.md | 16 ++++++++--------
1 file changed, 8 insertions(+), 8 deletions(-)
diff --git a/source/_posts/DeePMD_2026_05_19.md b/source/_posts/DeePMD_2026_05_19.md
index 649e59d..1f0f0c2 100644
--- a/source/_posts/DeePMD_2026_05_19.md
+++ b/source/_posts/DeePMD_2026_05_19.md
@@ -25,15 +25,15 @@ The research selected the Volmer step (H₃O⁺ + e⁻ + * → H* + H₂O), a ke
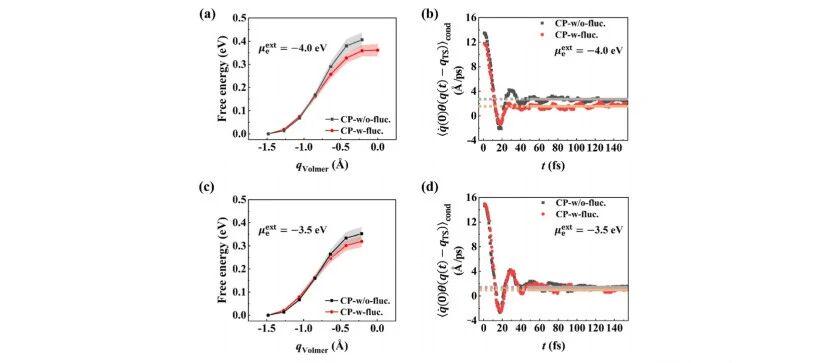 -Figure 1: (a,c) Free energy profiles along the defined reaction coordinate (RC); (b,d) Dynamic time-correlation function $\langle \dot{q}(0)\theta\left(q(t)-q\_\text{TS}\right) \rangle\_\text{cond}$ at different electrochemical potentials calculated by CP-w-fluc. and CP-w/o-fluc. methods. $\langle \cdot \rangle\_\text{cond}$ refers to the conditional average under the constraint $q(t=0)=q\_\text{TS}$. Orange and gray guidelines mark the plateau values of time correlation functions for the two methods. Filled error bands in (a,c) represent standard errors derived from 10 independent mean force simulations.
+*[Figure 1: (a,c) Free energy profiles along the defined reaction coordinate (RC); (b,d) Dynamic time-correlation function ⟨q̇(0)θ(q(t)−qTS)⟩cond at different electrochemical potentials calculated by CP-w-fluc. and CP-w/o-fluc. methods. ⟨⋅⟩cond refers to the conditional average under the constraint q(t=0)=qTS. Orange and gray guidelines mark the plateau values of time correlation functions for the two methods. Filled error bands in (a,c) represent standard errors derived from 10 independent mean force simulations.]*
-Under the same applied potential ($$\mu_\text{ex}$$ = −3.5 eV vs. vacuum), the transition state probability density obtained by CPwfluc. reaches 1.35×10⁻⁵/Å, while CPw/ofluc. only gives 3.38×10⁻⁶/Å, a four-fold difference. A similar trend is observed at −4.0 eV.
+Under the same applied potential (μex = −3.5 eV vs. vacuum), the transition state probability density obtained by CPwfluc. reaches 1.35×10−5/Å, while CPw/ofluc. only gives 3.38×10−6/Å, a four-fold difference. A similar trend is observed at −4.0 eV.
For the dynamic recrossing term, the plateau values of flux-side time correlation functions from CPwfluc. are generally lower, indicating more significant recrossing effects captured by this method. Combined results of the two terms are listed in Table 1.
-Figure 1: (a,c) Free energy profiles along the defined reaction coordinate (RC); (b,d) Dynamic time-correlation function $\langle \dot{q}(0)\theta\left(q(t)-q\_\text{TS}\right) \rangle\_\text{cond}$ at different electrochemical potentials calculated by CP-w-fluc. and CP-w/o-fluc. methods. $\langle \cdot \rangle\_\text{cond}$ refers to the conditional average under the constraint $q(t=0)=q\_\text{TS}$. Orange and gray guidelines mark the plateau values of time correlation functions for the two methods. Filled error bands in (a,c) represent standard errors derived from 10 independent mean force simulations.
+*[Figure 1: (a,c) Free energy profiles along the defined reaction coordinate (RC); (b,d) Dynamic time-correlation function ⟨q̇(0)θ(q(t)−qTS)⟩cond at different electrochemical potentials calculated by CP-w-fluc. and CP-w/o-fluc. methods. ⟨⋅⟩cond refers to the conditional average under the constraint q(t=0)=qTS. Orange and gray guidelines mark the plateau values of time correlation functions for the two methods. Filled error bands in (a,c) represent standard errors derived from 10 independent mean force simulations.]*
-Under the same applied potential ($$\mu_\text{ex}$$ = −3.5 eV vs. vacuum), the transition state probability density obtained by CPwfluc. reaches 1.35×10⁻⁵/Å, while CPw/ofluc. only gives 3.38×10⁻⁶/Å, a four-fold difference. A similar trend is observed at −4.0 eV.
+Under the same applied potential (μex = −3.5 eV vs. vacuum), the transition state probability density obtained by CPwfluc. reaches 1.35×10−5/Å, while CPw/ofluc. only gives 3.38×10−6/Å, a four-fold difference. A similar trend is observed at −4.0 eV.
For the dynamic recrossing term, the plateau values of flux-side time correlation functions from CPwfluc. are generally lower, indicating more significant recrossing effects captured by this method. Combined results of the two terms are listed in Table 1.
 -Table 1: Reaction Rate Constants ($$k_\text{BC}$$) at Different Electrochemical Potentials Calculated by CP-w/o-fluc. and CP-w-fluc. Methods
+*[Table 1: Reaction Rate Constants kBC at Different Electrochemical Potentials Calculated by CP-w/o-fluc. and CP-w-fluc. Methods]*
In short, under identical applied potentials, the rate constant predicted by CPwfluc. is roughly four times that of CPw/ofluc.
@@ -43,10 +43,10 @@ The observed discrepancy reveals the essence of the grand canonical ensemble: fl
Contrary to common perception, CPw/ofluc. is not more efficient. It requires repeated electron number iteration for convergence at every sampling step, resulting in heavier computational loads. In contrast, CPwfluc. evolves the total electron number $$N_e$$ as an independent dynamic sampling variable, featuring both stricter theories and higher computational efficiency.
-The two methods can be unified within extended ensemble dynamics via the coupling strength $$Q_\mu$$:
-- $$Q_\mu$$ → 0: Corresponds to CPw/ofluc. with instantaneous electron equilibrium and no fluctuation;
-- Moderate $$Q_\mu$$: Corresponds to CPwfluc. with dynamic fluctuations and accurate GCE sampling;
-- $$Q_\mu$$ → ∞: Corresponds to canonical ensemble sampling with fixed electron numbers.
+The two methods can be unified within extended ensemble dynamics via the coupling strength Qμ:
+- Qμ → 0: Corresponds to CPw/ofluc. with instantaneous electron equilibrium and no fluctuation;
+- Moderate Qμ: Corresponds to CPwfluc. with dynamic fluctuations and accurate GCE sampling;
+- Qμ → ∞: Corresponds to canonical ensemble sampling with fixed electron numbers.
## DP + ABACUS: Enabling Accessible Accurate Constant-Potential Simulations
@@ -54,7 +54,7 @@ Conventional ab initio molecular dynamics (AIMD) makes CPwfluc. computationally
All simulations were performed with neural network potentials trained by DP. The research team used the ABACUS first-principles software to generate DFT datasets and trained a DP-$$N_e$$ machine learning potential via the automatic DP-GEN workflow. This model incorporates the total electron number $$N_e$$ as well as its first and second partial derivatives. Leveraging numerical atomic orbital (NAO) basis sets, ABACUS efficiently handles large Pt/H₂O interface supercells containing hundreds of atoms. The DeePMD-kit trains deep learning force fields that precisely describe how atomic potential energy surfaces vary with electron numbers.
-The final DP model achieves root-mean-square errors (RMSE) of 0.6 meV/atom for energy and 54 meV/Å for force, reaching first-principles precision. Based on this potential, the team conducted long-time sampling with 300,000 steps per trajectory for systematic method comparison. The DP-$$N_e$$ model directly outputs $$\partial U/\partial N_e$$ and $$\partial^2 U/\partial N_e^2$$ via automatic differentiation, greatly accelerating electron number convergence in CPw/ofluc.
+The final DP model achieves root-mean-square errors (RMSE) of 0.6 meV/atom for energy and 54 meV/Å for force, reaching first-principles precision. Based on this potential, the team conducted long-time sampling with 300,000 steps per trajectory for systematic method comparison. The DP-Ne model directly outputs ∂U/∂Ne and ∂2U/∂Ne2 via automatic differentiation, greatly accelerating electron number convergence in CPw/ofluc.
## Conclusion
-Table 1: Reaction Rate Constants ($$k_\text{BC}$$) at Different Electrochemical Potentials Calculated by CP-w/o-fluc. and CP-w-fluc. Methods
+*[Table 1: Reaction Rate Constants kBC at Different Electrochemical Potentials Calculated by CP-w/o-fluc. and CP-w-fluc. Methods]*
In short, under identical applied potentials, the rate constant predicted by CPwfluc. is roughly four times that of CPw/ofluc.
@@ -43,10 +43,10 @@ The observed discrepancy reveals the essence of the grand canonical ensemble: fl
Contrary to common perception, CPw/ofluc. is not more efficient. It requires repeated electron number iteration for convergence at every sampling step, resulting in heavier computational loads. In contrast, CPwfluc. evolves the total electron number $$N_e$$ as an independent dynamic sampling variable, featuring both stricter theories and higher computational efficiency.
-The two methods can be unified within extended ensemble dynamics via the coupling strength $$Q_\mu$$:
-- $$Q_\mu$$ → 0: Corresponds to CPw/ofluc. with instantaneous electron equilibrium and no fluctuation;
-- Moderate $$Q_\mu$$: Corresponds to CPwfluc. with dynamic fluctuations and accurate GCE sampling;
-- $$Q_\mu$$ → ∞: Corresponds to canonical ensemble sampling with fixed electron numbers.
+The two methods can be unified within extended ensemble dynamics via the coupling strength Qμ:
+- Qμ → 0: Corresponds to CPw/ofluc. with instantaneous electron equilibrium and no fluctuation;
+- Moderate Qμ: Corresponds to CPwfluc. with dynamic fluctuations and accurate GCE sampling;
+- Qμ → ∞: Corresponds to canonical ensemble sampling with fixed electron numbers.
## DP + ABACUS: Enabling Accessible Accurate Constant-Potential Simulations
@@ -54,7 +54,7 @@ Conventional ab initio molecular dynamics (AIMD) makes CPwfluc. computationally
All simulations were performed with neural network potentials trained by DP. The research team used the ABACUS first-principles software to generate DFT datasets and trained a DP-$$N_e$$ machine learning potential via the automatic DP-GEN workflow. This model incorporates the total electron number $$N_e$$ as well as its first and second partial derivatives. Leveraging numerical atomic orbital (NAO) basis sets, ABACUS efficiently handles large Pt/H₂O interface supercells containing hundreds of atoms. The DeePMD-kit trains deep learning force fields that precisely describe how atomic potential energy surfaces vary with electron numbers.
-The final DP model achieves root-mean-square errors (RMSE) of 0.6 meV/atom for energy and 54 meV/Å for force, reaching first-principles precision. Based on this potential, the team conducted long-time sampling with 300,000 steps per trajectory for systematic method comparison. The DP-$$N_e$$ model directly outputs $$\partial U/\partial N_e$$ and $$\partial^2 U/\partial N_e^2$$ via automatic differentiation, greatly accelerating electron number convergence in CPw/ofluc.
+The final DP model achieves root-mean-square errors (RMSE) of 0.6 meV/atom for energy and 54 meV/Å for force, reaching first-principles precision. Based on this potential, the team conducted long-time sampling with 300,000 steps per trajectory for systematic method comparison. The DP-Ne model directly outputs ∂U/∂Ne and ∂2U/∂Ne2 via automatic differentiation, greatly accelerating electron number convergence in CPw/ofluc.
## Conclusion