scTenifoldNet is an R package for constructing and comparing single-cell gene regulatory networks (scGRNs) from single-cell RNA-seq (scRNA-seq) data collected under different conditions. It employs a machine-learning workflow combining principal component regression, tensor decomposition, and manifold alignment to accurately identify even subtly shifted gene expression programs.
Implementations in other languages are also available:
- Python: scTenifoldpy
- MATLAB: scGEAToolbox
scTenifoldNet is available on CRAN:
install.packages("scTenifoldNet")To install the development version from GitHub:
# install.packages("remotes")
remotes::install_github("cailab-tamu/scTenifoldNet")The scTenifoldNet() function orchestrates a six-step pipeline. Each step reports progress to the console via the cli package, with sample-level labels ([X] / [Y]) so that operations on each condition are clearly distinguishable.
| Step | Function | Description |
|---|---|---|
| 1 | scQC |
Quality control — filters cells by library size, outlier detection, minimum gene expression fraction, and mitochondrial read ratio |
| 2 | cpmNormalization |
Counts-per-million (CPM) normalization |
| 3 | makeNetworks |
Constructs gene regulatory networks from subsampled cells using principal component regression (pcNet) |
| 4 | tensorDecomposition |
CANDECOMP/PARAFAC (CP) tensor decomposition for network denoising |
| 5 | manifoldAlignment |
Non-linear manifold alignment of the two denoised networks |
| 6 | dRegulation |
Differential regulation testing via Box-Cox transformation and chi-square statistics |
Individual functions are exported and fully documented, allowing users to run or modify any step independently.
The required input is a raw counts matrix with genes as rows and cells (barcodes) as columns. Data should be unnormalized when using the main scTenifoldNet() function. The modular design of the package allows users to substitute custom preprocessing at any step.
scTenifoldNet() returns a list with three elements:
tensorNetworks— Weight-averaged denoised gene regulatory networks after CP tensor decomposition, containing:X: The network for sample X (sparse matrix of classdgCMatrix).Y: The network for sample Y (sparse matrix of classdgCMatrix).
manifoldAlignment— A data frame of low-dimensional features from the non-linear manifold alignment, with 2 × n genes rows and d columns (default d = 30).diffRegulation— A data frame with six columns:gene: Gene identifier.distance: Euclidean distance between the gene's coordinates in the two conditions.Z: Z-score after Box-Cox power transformation.FC: Fold change with respect to the expectation.p.value: P-value from the chi-square distribution with one degree of freedom.p.adj: Adjusted p-value (Benjamini & Hochberg FDR correction).
Running time is largely determined by the network construction step and scales with the number of cells and genes. Representative benchmarks:
| Cells | Genes | Time | Memory |
|---|---|---|---|
| 300 | 1,000 | 3.5 min | 0.4 GB |
| 1,000 | 1,000 | 4.3 min | 0.4 GB |
| 1,000 | 5,000 | 2 h 52 min | 9.2 GB |
| 2,500 | 5,000 | 2 h 55 min | 9.5 GB |
| 5,000 | 5,000 | 3 h 9 min | 10.1 GB |
| 5,000 | 7,500 | 3 h 10 min | 21.8 GB |
| 7,500 | 5,000 | 10 h 16 min | 10.6 GB |
| 7,500 | 7,500 | 10 h 16 min | 22.6 GB |
We create a sparse count matrix of 2,000 cells and 100 genes drawn from a negative binomial distribution (~67 % zeros). The last ten genes are prefixed with mt- to simulate mitochondrial genes.
library(scTenifoldNet)
nCells <- 2000
nGenes <- 100
set.seed(1)
X <- matrix(
rnbinom(n = nGenes * nCells, size = 20, prob = 0.98),
ncol = nCells
)
rownames(X) <- c(paste0("ng", 1:90), paste0("mt-", 1:10))Genes 10, 2, and 3 are replaced with the expression profiles of genes 50, 11, and 5, respectively.
Y <- X
Y[10, ] <- Y[50, ]
Y[2, ] <- Y[11, ]
Y[3, ] <- Y[5, ]Under H₀ (no differential regulation) we supply the same matrix for both conditions. Under H_A we supply the control and perturbed matrices.
outputH0 <- scTenifoldNet(X = X, Y = X,
nc_nNet = 10, nc_nCells = 500,
td_K = 3, qc_minLibSize = 30)
outputHA <- scTenifoldNet(X = X, Y = Y,
nc_nNet = 10, nc_nCells = 500,
td_K = 3, qc_minLibSize = 30)The denoised networks are stored as sparse matrices that can be converted to igraph objects for downstream topological analysis:
igraph::graph_from_adjacency_matrix(outputHA$tensorNetworks$X, weighted = TRUE)
igraph::graph_from_adjacency_matrix(outputHA$tensorNetworks$Y, weighted = TRUE)Under H₀, no genes reach significance (FDR < 0.1). Under H_A, the six genes involved in the perturbation are correctly identified:
head(outputH0$diffRegulation, n = 6)
# gene distance Z FC p.value p.adj
# 59 ng59 1.138659e-15 2.497859 4.338493 0.03725989 0.6411916
# 32 ng32 9.970451e-16 2.081719 3.326448 0.06817394 0.6411916
# 72 ng72 9.452170e-16 1.917581 2.989608 0.08380044 0.6411916
# 89 ng89 8.924023e-16 1.742758 2.664849 0.10258758 0.6411916
# 12 ng12 8.851226e-16 1.718018 2.621550 0.10542145 0.6411916
# 17 ng17 8.784453e-16 1.695183 2.582145 0.10807510 0.6411916
head(outputHA$diffRegulation, n = 6)
# gene distance Z FC p.value p.adj
# 2 ng2 0.023526702 2.762449 12.193413 0.0004795855 0.02414332
# 50 ng50 0.023514429 2.761550 12.180695 0.0004828665 0.02414332
# 11 ng11 0.022443941 2.681598 11.096894 0.0008647241 0.02882414
# 3 ng3 0.020263415 2.508478 9.045415 0.0026335445 0.06583861
# 10 ng10 0.019194561 2.417929 8.116328 0.0043868326 0.07711821
# 5 ng5 0.019079975 2.407977 8.019712 0.0046270923 0.07711821Quantile-quantile plots provide a clear summary. Genes with FDR < 0.1 are highlighted in red.
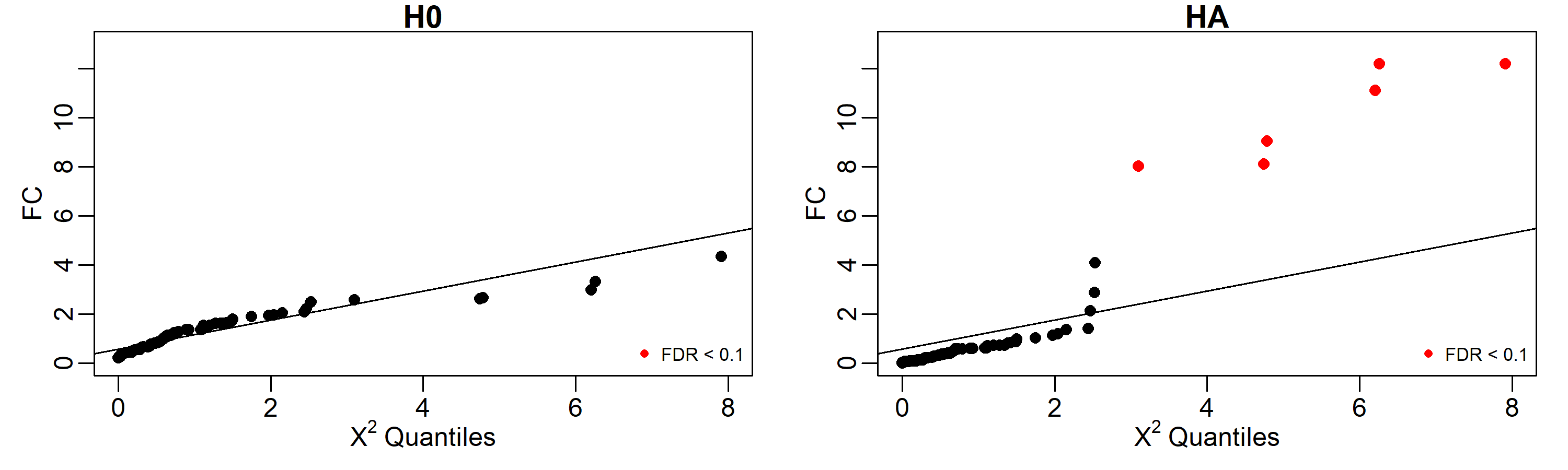
par(mfrow = c(2, 1), mar = c(3, 3, 1, 1), mgp = c(1.5, 0.5, 0))
set.seed(1)
qChisq <- rchisq(100, 1)
geneColor <- rev(ifelse(outputH0$diffRegulation$p.adj < 0.1, "red", "black"))
qqplot(qChisq, outputH0$diffRegulation$FC, pch = 16, main = expression(H[0]),
col = geneColor, xlab = expression(chi^2 ~ Quantiles), ylab = "FC",
xlim = c(0, 8), ylim = c(0, 13))
qqline(qChisq)
legend("bottomright", legend = "FDR < 0.1", pch = 16, col = "red", bty = "n", cex = 0.7)
geneColor <- rev(ifelse(outputHA$diffRegulation$p.adj < 0.1, "red", "black"))
qqplot(qChisq, outputHA$diffRegulation$FC, pch = 16, main = expression(H[A]),
col = geneColor, xlab = expression(chi^2 ~ Quantiles), ylab = "FC",
xlim = c(0, 8), ylim = c(0, 13))
qqline(qChisq)
legend("bottomright", legend = "FDR < 0.1", pch = 16, col = "red", bty = "n", cex = 0.7)If you use scTenifoldNet in your research, please cite:
Osorio, D., Zhong, Y., Li, G., Huang, J. Z., & Cai, J. J. (2020). scTenifoldNet: A Machine Learning Workflow for Constructing and Comparing Transcriptome-wide Gene Regulatory Networks from Single-Cell Data. Patterns, 1(9), 100139. doi:10.1016/j.patter.2020.100139
BibTeX:
@Article{osorio2020sctenifoldnet,
title = {scTenifoldNet: A Machine Learning Workflow for Constructing and
Comparing Transcriptome-wide Gene Regulatory Networks from
Single-Cell Data},
author = {Daniel Osorio and Yan Zhong and Guanxun Li and Jianhua Z. Huang
and James J. Cai},
journal = {Patterns},
year = {2020},
volume = {1},
number = {9},
pages = {100139},
issn = {2666-3899},
doi = {10.1016/j.patter.2020.100139},
}© The Texas A&M University System. All rights reserved.