The purpose of these scripts is to quantify and identify read alignment hits, sequence, organism and taxonomy information from Binary Alignment Maps files (BAM) into a csv file
Steps: First thing is that you will need to create a conda environment and install Entrez in these instructions https://bioconda.github.io/recipes/entrez-direct/README.html (Just do the install and update commands)
Download both scripts (python and shell) to your directory. The two scripts work together for CSV conversion.
In order for the scripts to function correctly, the BAM files in your directory must be named in the form of binomial nomenclature of the particular organism. The format of the file name must look like this ex. (NEW_Myotis__lucifugus.bam) The keywords of "NEW_" and "__" are meant to be recognized and replaced when converting into a CSV file.
Before running the script, it is important that you edit the directory path to yours specifically. Another aspect that needs to be edited is depended on what organism domain (Eukaryota, Bacteria, Archaea) you are working with. The keywords are specifed in the script and must be changed when dealing with a different organism domain.
Once prepared and edits are made the script is ready to go. (The shell script is the only script that has to be executed)
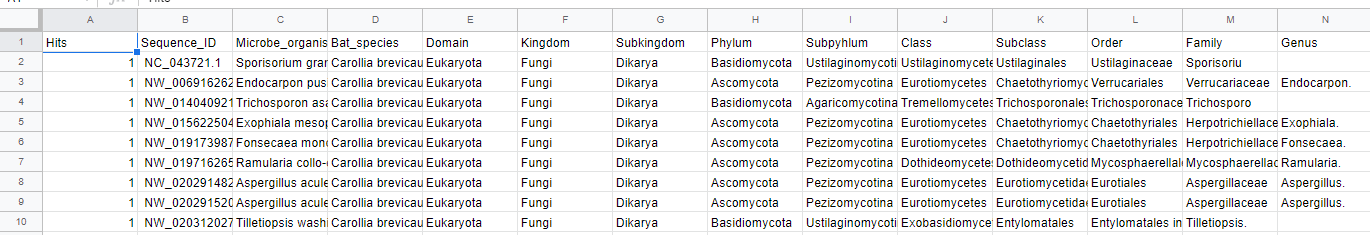
An example of the CSV output file